

BREAKING NEWS
Site Under Development, Content Population and SEO, Soft Launch 1st January 2020
Polycystic kidney disease (PKD) is a hereditary condition in which the kidneys develop multiple cysts. This leads to renal enlargement, distortion of the normal structure of the kidneys and functional impairment.
The result may be chronic kidney disease, progressing to end-stage kidney disease (ESRD) requiring dialysis or kidney transplantation.
It affects both genders and all ethnic groups but leads to chronic kidney disease in males and in women who had three or more pregnancies and who have high blood pressure.
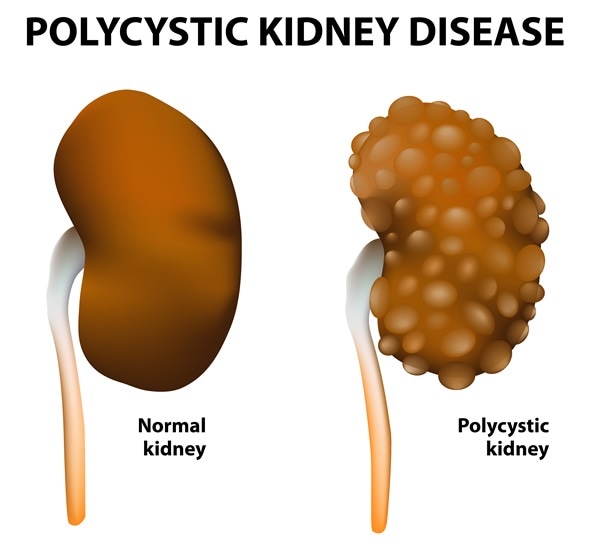
PKD often presents with pain in the back and the sides of the ribcage and headaches, while hematuria is sometimes present. In autosomal recessive PKD, enlarged kidneys, respiratory distress and growth failure in infants may be the presenting symptoms.
PKD may be autosomal dominant or recessive. Autosomal dominant PKD occurs in up to one in 400 people but the autosomal recessive type occurs in one in 20 000 – 40 000 people.
Credit: PKD Foundation
PKD is the result of a gene defect, which is usually present in the parent and is passed on to the offspring. At least three mutations have been found to be associated with PKD, of which two are found in the autosomal dominant type.
The most common form of PKD is the autosomal dominant type. It usually starts between 30 and 50 years, but symptoms start only when the cysts have reached a size of 0.5 cm or more. In the recessive form, cysts form in infancy or even in utero.
PKD is diagnosed on the basis of imaging tests and genetic testing. Imaging the kidneys involves ultrasound, CT scans or MRI scans. A scan which shows two or more cysts before the age of 30, for instance, combined with a family history of PKD, may lead to a diagnosis of the condition.
Genetic testing is carried out on saliva or blood and looks for the presence of specific gene mutations. This helps with diagnosis as well as predicting the chances of passing on the disease. Prenatal genetic testing can identify the presence of the gene in fetuses in utero. However, genetic testing fails to predict the time of onset of symptoms or their severity.
PKD has no cure and treatment is directed towards symptom control and achieving a longer life. This may involve cyst removal to relieve pain, bringing down the blood pressure, lifestyle changes and monitoring for heart valve complications or brain aneurysms. For instance, diet is important in preventing some complications, mostly due to high blood pressure and so a low-sodium diet with high intake of vegetables is often recommended, with more poultry, fish or dairy products for protein.
In the autosomal recessive form of PKD, the kidneys may be so large that they prevent normal breathing. In such cases, nephrectomy is unavoidable and such children must receive a kidney transplant or dialysis. Proper growth is ensured by dietary guidelines, sometimes in conjunction with growth hormone treatment for severe growth arrest.
Respiratory arrest is always a possibility in infants with the severe form of the disease and ventilator support is often required, as well as cardiopulmonary resuscitation in emergency situations. Kidney function is replaced by dialysis, usually peritoneal, but also hemodialysis in some cases. Control of urinary infections, hypertension and measures to slow the progression of liver disease is required. Combined liver and kidney transplantation is necessary in some cases.